
The biological complexity of rare genetic disorders often creates a formidable barrier that traditional pharmaceutical models struggle to overcome, leaving thousands of families in a state of clinical limbo. While a single rare disease might only affect a small cohort of individuals, the collective burden of the thousands of known orphan conditions impacts millions of people globally, highlighting a systemic gap in modern healthcare. Historically, the astronomical costs associated with research and development, combined with the limited commercial return from small patient populations, have deterred large-scale private investment. This economic reality means that many life-threatening conditions remain under-researched, with patients relying on repurposed medications that only address superficial symptoms rather than the root genetic cause. However, the landscape is shifting as innovative funding models and precision medicine begin to prioritize the unique needs of these underserved communities. By focusing on the shared molecular mechanisms of these disorders, researchers are finding that breakthroughs in one area can often illuminate the path for others, creating a ripple effect of innovation across the entire field of genetics.
To bridge the financial and scientific divide, patient-led organizations have emerged as the primary catalysts for modern medical breakthroughs in the rare disease sector. Groups such as the AT Society and the Cystic Fibrosis Trust have moved beyond traditional advocacy to become sophisticated venture philanthropists, funding high-risk, early-stage academic research that the private sector typically deems too speculative. These organizations provide the essential “seed” capital that allows university researchers to develop proof-of-concept data, which is often the prerequisite for attracting larger institutional or industrial interest. By maintaining a laser focus on the patient experience, these groups ensure that research agendas are aligned with the most pressing clinical needs, such as maintaining mobility or reducing pulmonary exacerbations. This collaborative ecosystem between families, scientists, and clinicians has successfully transformed the trajectory of several conditions, proving that dedicated advocacy can effectively de-risk drug development and pave the way for commercial viability and widespread clinical access.
Navigating the Complexity of Ataxia Telangiectasia
Ataxia telangiectasia is a devastating multi-system disorder that serves as a stark reminder of how a single genetic mutation can disrupt the delicate harmony of the human body. Children born with this condition often appear perfectly healthy during infancy, but as they reach early childhood, the first signs of neurological decline begin to emerge through a subtle lack of motor coordination. This progressive loss of balance, known as ataxia, is merely the visible tip of a much deeper physiological iceberg that eventually impacts the immune, respiratory, and endocrine systems. By the time many of these children reach the age of ten, the rapid progression of neurodegeneration often necessitates the use of a wheelchair, fundamentally altering their independence and quality of life. The systemic nature of the disease means that every milestone is met with the shadow of chronic fatigue and physical frailty, creating an urgent need for interventions that can stabilize these functions before irreversible damage occurs.
Beyond the immediate challenges of physical mobility, the underlying biological instability of the condition introduces a heightened risk of life-threatening complications, particularly regarding immune function and oncology. Approximately two-thirds of individuals living with the disorder suffer from a compromised immune system, leaving them perpetually vulnerable to severe respiratory infections and chronic sinus issues. Even more concerning is the statistical reality that roughly twenty-five percent of patients will develop some form of malignancy, ranging from pediatric leukemias to aggressive solid tumors in early adulthood. This high predisposition to cancer stems from the body’s inability to manage cellular stress and DNA repair, making traditional treatments like radiation therapy extremely dangerous for this specific population. Consequently, the medical community is shifting its focus from palliative care toward genetic strategies that aim to restore cellular stability, hoping to mitigate these secondary risks and provide a more secure future for those affected.
Genetic Obstacles and the Role of the ATM Protein
At the heart of this complex clinical picture is a mutation in the ATM gene, which serves as a critical master regulator for maintaining the integrity of the human genome. The ATM protein produced by this gene acts as a cellular sentinel, detecting DNA double-strand breaks and orchestrating the complex repair processes necessary for healthy cell division. When this protein is absent or dysfunctional, the cell loses its ability to correct genetic errors, leading to the accumulation of mutations and the eventual death of neurons in the cerebellum. The sheer size of the ATM gene, which encompasses over 150 kilobases of DNA, presents a monumental challenge for modern gene therapy. Most conventional viral vectors, which are the standard delivery vehicles for genetic medicine, simply do not have the carrying capacity to transport such a massive sequence into a patient’s cells. This physical limitation has historically stalled progress, as researchers could not use the same tools that have been successful in treating smaller genetic defects.
The technical bottleneck created by the size of the ATM gene has forced the scientific community to pivot toward non-viral delivery systems and alternative molecular strategies. Instead of trying to squeeze a massive gene into a tiny viral shell, researchers are exploring the use of synthetic materials and advanced nanotechnology to bypass traditional constraints. This shift is not merely a workaround; it represents a fundamental change in how genetic material is handled and protected as it moves through the bloodstream and across biological barriers like the blood-brain barrier. Because the neurological symptoms of the disease are the most debilitating, any successful therapy must be able to reach the deep tissues of the central nervous system effectively. Solving the delivery problem for the ATM gene could potentially unlock treatments for a wide range of other “large-gene” disorders that have previously been considered untreatable, marking a significant milestone in the evolution of genomic medicine and therapeutic delivery.
Breakthroughs in Delivery and Gene Correction
Recent advancements at the University of Nottingham have introduced a promising new frontier in the treatment of rare diseases through the development of peptide-based nanoparticles. These engineered particles are designed to act as highly specialized transport vehicles, capable of shielding sensitive genetic payloads from the body’s immune system while navigating directly to the targeted cells. Unlike viral delivery methods, these non-viral nanoparticles can be engineered to carry much larger genetic sequences, finally making the delivery of the ATM gene a technical possibility. Furthermore, these synthetic platforms offer the advantage of being “redosable,” meaning they do not trigger the permanent immune resistance that often prevents patients from receiving multiple rounds of viral gene therapy. This flexibility is vital for treating chronic, progressive conditions where sustained therapeutic levels are necessary to halt the ongoing degeneration of the nervous system and other vital organs.
These nanoparticle platforms are not limited to carrying a full replacement gene; they are also being utilized to deliver sophisticated gene-editing tools and antisense oligonucleotides. This multi-pronged approach allows clinicians to tailor the treatment to the specific needs of the patient, whether that involves correcting a point mutation or modulating the way a gene is expressed. For instance, antisense oligonucleotides can be used to “skip” over faulty sections of genetic code, allowing the cell to produce a partially functional protein that may be enough to stop disease progression. By providing a versatile delivery system that can be adapted for various molecular strategies, researchers are moving closer to a reality where a single technological framework can treat multiple classes of mutations. This development offers a beacon of hope for families, as it suggests that the time between laboratory discovery and clinical application could be significantly shortened through the use of standardized, safe delivery platforms.
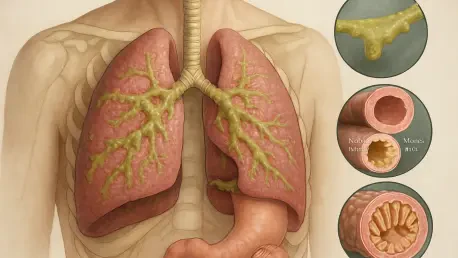
Understanding the Impact of Cystic Fibrosis
Cystic fibrosis stands as one of the most well-known rare genetic conditions, yet its underlying pathophysiology remains a complex challenge for those living with the disease and the clinicians who treat them. The disorder is caused by mutations in the CFTR gene, which is responsible for regulating the movement of salt and water across cell membranes in the lungs, pancreas, and other organs. When this gene is defective, the result is the production of abnormally thick and sticky mucus that clogs vital pathways, leading to a cycle of chronic infection and inflammation. In the respiratory system, this buildup creates a breeding ground for bacteria, resulting in permanent lung damage and a gradual decline in pulmonary function over time. While modern care has greatly extended life expectancy, the daily burden of clearing airways and managing digestive complications remains a constant struggle for thousands of individuals within the United Kingdom and beyond.
The genetic landscape of the condition is particularly noteworthy because of its prevalence as an autosomal recessive trait, meaning that many people carry the mutation without ever showing symptoms. In the UK, it is estimated that one in twenty-five people is a carrier of a faulty CFTR gene, often discovering this fact only when a child is born with the condition. This high carrier frequency explains why the disease remains relatively common despite its classification as a rare disorder. For decades, the medical community could do little more than manage the symptoms through aggressive antibiotic use and physical therapy to manually clear the lungs. However, the deep understanding of the CFTR protein gained through years of intensive research has finally led to a new era of treatment. Instead of just reacting to the damage caused by the mucus, new therapeutic strategies are now focusing on the protein itself, attempting to fix its function at the cellular level to prevent the mucus from ever becoming thick and obstructive.
The Transformation of Care Through CFTR Modulators
The introduction of CFTR modulators has represented one of the most significant triumphs of precision medicine in the modern era, fundamentally changing the prognosis for the vast majority of patients. These small-molecule drugs do not just treat the symptoms of the disease; they interact directly with the defective CFTR protein to improve its stability and function. Some of these molecules, known as correctors, help the protein fold into its proper three-dimensional shape so that it can successfully reach the cell surface instead of being degraded by the cell’s internal quality control. Others, called potentiators, act as a “key” that keeps the protein’s chloride channel open for longer periods, allowing for a more natural flow of salt and water. The synergy of these different types of modulators has led to dramatic improvements in lung function, weight gain, and overall energy levels for approximately ninety percent of the patient population.
This clinical revolution was made possible by the application of high-throughput screening technologies, which allowed researchers to test hundreds of thousands of chemical compounds to find those with the specific ability to interact with the CFTR protein. This data-driven approach bypassed much of the traditional guesswork involved in drug discovery, leading to the rapid development and approval of transformative therapies like Kaftrio. For those who respond to these treatments, the daily reality of the disease has been shifted from a constant battle for breath to a more manageable chronic condition. The success of these modulators serves as a powerful proof of concept for the entire field of rare disease research, demonstrating that when the underlying molecular defect is understood, it is possible to develop highly effective, targeted therapies that can change the trajectory of a previously life-shortening illness.
Closing the Gap for All Patients
Despite the remarkable success of modulator therapies, there remains a critical “final ten percent” of the cystic fibrosis population whose specific genetic mutations do not respond to existing small-molecule drugs. These individuals often have “nonsense” mutations or other rare variants that result in no protein being produced at all, leaving them with no target for current modulators to act upon. For these patients, the daily routine still involves hours of nebulized treatments, frequent hospitalizations for intravenous antibiotics, and the looming possibility of a lung transplant. The scientific community has recognized this disparity as an urgent call to action, shifting its focus toward developing “mutation-agnostic” therapies that could benefit every patient regardless of their specific genetic code. This includes the exploration of mRNA therapies, which provide the cell with the instructions to make a healthy protein, bypassing the faulty gene entirely.
The drive to leave no patient behind has fostered a new wave of innovation that mirrors the work being done in other rare diseases like ataxia telangiectasia. By leveraging the same nanoparticle delivery systems and gene-editing technologies, researchers are working to find permanent or long-term solutions for this underserved group. The goal is to move beyond the need for daily medication and instead provide a durable correction that allows the body to function normally on its own. This ongoing effort is characterized by a high degree of international collaboration, as the small size of this remaining patient cohort requires a global approach to clinical trials and data sharing. As these new technologies advance, the focus is not just on matching the success of current modulators, but on exceeding it by providing a universal solution that addresses the root cause of the disease for every single person affected by it.
The Future of Personalized and Platform-Based Medicine
The evolution of treatments for these conditions underscores a broader shift toward a “platform-based” approach in medicine, where the delivery technology is just as important as the therapeutic agent itself. By developing standardized, non-viral delivery systems like the peptide nanoparticles used in neurological research, scientists are creating a toolkit that can be rapidly adapted to a wide variety of genetic disorders. This means that once a delivery vehicle is proven safe and effective for one condition, it can be “re-loaded” with different genetic instructions to treat another, drastically reducing the time and cost associated with developing new therapies from scratch. This transition from bespoke, one-off treatments to versatile therapeutic platforms is essential for addressing the thousands of rare diseases that currently lack any effective intervention. It allows the medical community to treat these conditions as a collective challenge rather than a series of isolated, unconnected problems.
Furthermore, the integration of personalized medicine into standard care is ensuring that treatments are increasingly tailored to the unique biological profile of each individual. As genomic sequencing becomes more accessible and detailed, clinicians can identify the exact molecular drivers of a patient’s disease and select the most effective combination of therapies from the outset. This precision reduces the “trial and error” phase of treatment, minimizing side effects and maximizing the therapeutic benefit for the patient. The success seen in restoring protein function in cystic fibrosis is now serving as a blueprint for other fields, encouraging a shift toward early intervention and disease modification. By focusing on the cellular roots of health and disease, the next generation of medical treatments will likely be defined by their ability to not only extend life but to preserve the highest possible level of physical and cognitive function throughout a person’s lifetime.
Advocacy as a Catalyst for Scientific Discovery
The journey from understanding the genetic basis of rare diseases to delivering life-changing treatments has been fueled by the unwavering commitment of patient advocacy groups. These organizations have fundamentally changed the rules of engagement in medical research by proving that a dedicated community can overcome the economic hurdles that once stalled progress in the orphan disease space. Moving forward, it is essential for the global healthcare community to maintain this momentum by continuing to support the collaborative models that have proven so successful. This involves not only funding research but also ensuring that regulatory frameworks are flexible enough to accommodate the unique challenges of small-batch, personalized genetic therapies. The path forward requires a sustained commitment to transparency, data sharing, and international cooperation to ensure that the breakthroughs of today become the standard of care for every patient tomorrow.
As the focus shifts toward even more advanced solutions, such as in vivo gene editing and systemic protein restoration, the primary objective must remain the enhancement of patient dignity and independence. The technical ability to correct a genetic defect is only truly valuable if it translates into a child being able to walk, a young adult being able to breathe without assistance, and families being freed from the constant anxiety of a progressive illness. Future efforts should prioritize the expansion of clinical trials to include more diverse genetic backgrounds and the development of cost-effective manufacturing processes to ensure these therapies are accessible to everyone, not just those in the wealthiest regions. By staying focused on these actionable goals and building upon the successful foundation of platform-based medicine and patient-led innovation, the dream of providing a cure for every rare disease is no longer a distant hope but a tangible objective that is being realized with each passing year.